

BioMoleculeAlign
BioMoleculeAlign[ref,biomol]
returns a copy of biomol that has been aligned with reference biomolecule ref.
BioMoleculeAlign[ref,biomol,mapping]
uses the supplied mapping to determine which chains to include in the alignment.
BioMoleculeAlign[ref,biomol,mapping,prop]
aligns the biomolecules and returns the property prop of the alignment.
Details
- BioMoleculeAlign is used to structurally align two peptide or nucleic acid molecules in space.
- BioMoleculeAlign uses a structure-based sequence-independent algorithm to align 3D structures by optimizing the template-modeling score objective function, or TM-score. It is often used when comparing a predicted structure to an experimentally determined structure.
- The TM-score for a given alignment is defined by
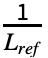 ∑i
∑i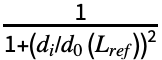 , where Lref is the length of the reference structure, di is the distance between backbone atoms for the ith aligned residue, and d0(Lref)=1.24
, where Lref is the length of the reference structure, di is the distance between backbone atoms for the ith aligned residue, and d0(Lref)=1.24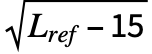 -1.8 is a distance scale to normalize the result. The alignment that maximizes the TM-score is returned.
-1.8 is a distance scale to normalize the result. The alignment that maximizes the TM-score is returned. - The TM-score lies between 0 and 1, where 1 indicates a perfect match. A TM-score below 0.2 is considered to be from random chance, while a score over 0.5 indicates a roughly the same folding.
- mapping should be either Automatic or a rule between chain labels in ref and biomol, e.g. {"A","B"}{"C","D"}. When a rule is given, only the specified chains will be considered in the alignment.
- prop can be one of the following:
-
"BioMolecule" the aligned biomolecule "Transformation" the transformation function used "RMSError" the alignment root mean squared error "TMScore" the template-modeling score for the alignment "AlignedResidueCount" the number of residues aligned "SequenceIdentity" fraction of aligned residues that are identitical "SequenceCharacterAlignment" a visual representation of the aligned sequences All an Association with all properties
Examples
open all close allBasic Examples (3)
Create a biomolecule based on an experimental crystal structure:
experimental = Import["ExampleData/1PPT.pdb"]Now take the peptide sequence from that biomolecule and fold it:
predicted = BioMolecule[First[experimental["BioSequences"]]]The predicted structure's 3D coordinates are unrelated to the experimental structure:
BioMoleculePlot3D[{experimental, predicted}]Now align the predicted structure with the experimental structure and show them in 3D:
BioMoleculePlot3D[{experimental, BioMoleculeAlign[experimental, predicted]}]Align a smaller peptide to a specific chain in a larger peptide:
bm1 = BioMolecule[«1»];
bm2 = BioMolecule[«1»];
BioMoleculePlot3D[{bm1, BioMoleculeAlign[bm1, bm2, "B3" -> "A"]}]Align the smaller peptide to a different chain:
BioMoleculePlot3D[{bm1, BioMoleculeAlign[bm1, bm2, "A2" -> "A"]}]BioMoleculeAlign can align peptides with dissimilar sequences but similar 3D structures:
bm1 = BioMolecule[ExternalIdentifier["PDBStructureID", "101M"]];
bm2 = BioMolecule[ExternalIdentifier["PDBStructureID", "1AZI"]];
alignmentData = BioMoleculeAlign[bm1, bm2, Automatic, All];
BioMoleculePlot3D[{bm1, alignmentData["BioMolecule"]}]The structures are aligned very well as shown by the low RMSD error and high TM-score:
alignmentData[[{"RMSError", "TMScore"}]]Nearly all the amino acid residues participate in the alignment:
alignmentData["SequenceCharacterAlignment"]But there are significant differences in the two sequences:
SequenceAlignment[First[bm1["BioSequences"]], First[bm2["BioSequences"]]]Scope (2)
Align an RNA strand to the RNA portion of an RNA-peptide complex:
ref = BioMolecule[«1»];
rna = BioMolecule[«1»];
aligned = BioMoleculeAlign[ref, rna]BioMoleculePlot3D[{ref, aligned}]Compare the structures of Cyclin-dependent kinase 2 from two different organisms from AlphaFold Database:
bm1 = ServiceExecute["AlphaFoldDatabase", "BioMolecule", {"UniProtID" -> "A0A2K6V5L6"}];
bm2 = ServiceExecute["AlphaFoldDatabase", "BioMolecule", {"UniProtID" -> "A0A2K6GY84"}];
bma = BioMoleculeAlign[bm1, bm2];
BioMoleculePlot3D[{bm1, bma}, PlotLegends -> {"BioMolecule 1", "Aligned BioMolecule 2"}]Neat Examples (1)
Align two large biomolecular assemblies:
bm1 = BioMolecule[ExternalIdentifier["PDBStructureID", "1PV8"]]["Assemblies"][[1]];
bm2 = BioMolecule[ExternalIdentifier["PDBStructureID", "1E51"]]["Assemblies"][[1]];
bma = BioMoleculeAlign[bm1, bm2]colors = {RGBColor[0.6666666666666666, 0.6666666666666666, 0.6666666666666666], RGBColor[7/9, 1/3, 1/3]};
BioMoleculePlot3D[{Style[bm1, RGBColor[0.6666666666666666, 0.6666666666666666, 0.6666666666666666]], Style[bma, RGBColor[7/9, 1/3, 1/3]]}, PlotLegends -> {"BioMolecule 1", "Aligned BioMolecule 2"}]Text
Wolfram Research (2025), BioMoleculeAlign, Wolfram Language function, https://reference.wolfram.com/language/ref/BioMoleculeAlign.html.
CMS
Wolfram Language. 2025. "BioMoleculeAlign." Wolfram Language & System Documentation Center. Wolfram Research. https://reference.wolfram.com/language/ref/BioMoleculeAlign.html.
APA
Wolfram Language. (2025). BioMoleculeAlign. Wolfram Language & System Documentation Center. Retrieved from https://reference.wolfram.com/language/ref/BioMoleculeAlign.html